Summary of recent progress in the field of cancer
Summary of recent progress in the field of cancer
November 15, 2017 Source: WuXi PharmaTech
Window._bd_share_config={ "common":{ "bdSnsKey":{ },"bdText":"","bdMini":"2","bdMiniList":false,"bdPic":"","bdStyle":" 0","bdSize":"16"},"share":{ }};with(document)0[(getElementsByTagName('head')[0]||body).appendChild(createElement('script')) .src='http://bdimg.share.baidu.com/static/api/js/share.js?v=89860593.js?cdnversion='+~(-new Date()/36e5)];1. GSK antibody drug conjugate new drug obtained FDA breakthrough therapy
Recently, GlaxoSmithKline (GSK) announced that the US FDA has granted its breakthrough drug therapy conjugate GSK2857916 breakthrough therapy for the treatment of refractory multiple myeloma.

Multiple myeloma is a type of blood cancer caused by malignant plasma cells. Normally plasma cells can make antibodies against pathogens, however, once they become cancerous, growth is uncontrolled. Clinically, if multiple plasmacytomas appear at the same time, it is called multiple myeloma. Patients with this disease will have a series of symptoms such as reduced blood count, infection, bone problems, and kidney problems. The median survival of early patients can be more than 5 years, and the median survival of patients with advanced disease will be significantly reduced.
GSK2857916, brought by GSK, is an antibody drug conjugate derived from a humanized BCMA monoclonal antibody conjugated to the cytotoxic drug MMAF (monomethyl auristatin-F) via a linker. Currently, it is in the early stages of clinical development to treat relapsed/refractory multiple myeloma, as well as other malignant hematological cancers that express BCMA.
The potential of this new drug has been recognized by many regulatory agencies. In October of this year, EMA issued a PRIME certification. In early November, the US FDA immediately awarded it a breakthrough therapy. These two determinations are based on a publicly labelled, dose escalation Phase 1 clinical trial. The specific data of the trial will be announced at the American Society of Hematology (ASH) annual meeting on December 11 this year.
Dr. Axel Hoos, Senior Vice President of Oncology Research and Development at GSK, said: "GSK's Oncology Research Department is focused on developing new drugs with revolutionary potential for patients. We are pleased to see that the antibody conjugates under development are the first to be acquired simultaneously. FDA breakthrough treatment, and RIM's PRIME-recognized anti-BCMA therapy. GSK plans to advance this single-agent and combination therapy clinical trial of this potential therapy to further investigate how GSK2857916 will benefit patients with multiple myeloma The data we see in monotherapy supports its revolutionary potential, and we look forward to working with regulators to advance our development projects."
2. 3-year data show that Opdivo significantly prolongs survival in patients with kidney cancer
Recently, Bristol-Myers Squibb (BMS) announced the results of its Phase 3 clinical trial of CheckMate-025 at the 16th International Kidney Cancer Symposium in Miami, Florida, USA: Compared to existing therapies, Opdivo (nivolumab) provides excellent 3-year survival for previously treated patients with advanced renal cell carcinoma (RCC). This is the first time that PD-1 mAb has shown a 3-year long-lasting survival rate in this type of patient.

Renal cell carcinoma is the most common type of kidney cancer in adults, and about 100,000 people die every year from RCC worldwide. Globally, the 5-year survival rate for patients diagnosed with metastatic or advanced renal cancer is only 12.1%.
CheckMate-025 is an open, randomized phase 3 clinical study of Opdivo that compares existing treatments with patients with advanced renal cell carcinoma who have previously received anti-angiogenic therapy. The primary clinical endpoint of this study was overall survival (OS). Secondary endpoints included objective response rate (ORR), progression-free survival (PFS), quality of life (QoL), and safety. A total of 803 patients were enrolled in the study, of which 406 were enrolled in Opdivo and the remaining 397 were treated with existing therapies until disease progression or unacceptable toxicity.
The results showed that the median OS was 25.8 months in patients treated with Opdivo, compared with 19.7 months in the current therapy (HR 0.74; 95.45% CI: 0.63 to 0.88; p: 0.0005). Opdivo's three-year OS rate is 39%, compared to 30% for existing therapies. The study did not find new safety signals, and the data showed safety consistent with the 2-year results.
At 36 months, Opdivo showed a secondary clinical endpoint ORR of 26%, compared with 5% of current therapies (95% CI: 3.82 to 10.06) and a median duration of 12.3 for Opdivo and existing therapies, respectively. Months (95% CI: 9.1 to 18.2) and 12 months (95% CI: 6.4 to 21.7). In another clinical secondary endpoint, the median PFS was 4.2 months for Opdivo and 4.5 months for the current therapy (HR 0.85; 95% CI: 0.73 to 0.99; P: 0.0371).
Dr. Arvin Yang, head of the BMS melanoma and genitourinary cancer research and development department, said: "CheckMate-025 data strengthens the care standard for Opdivo in previously treated patients with renal cell carcinoma. The three-year survival data shows that Existing therapies provide patients with an improvement of 6 months of survival. Based on this unprecedented two-year overall survival data, this is the first three-year overall survival rate for PD-1 mAbs in patients with advanced RCC. Verifies our commitment and confidence to improve patient survival in the most common types of kidney cancer in adults worldwide."
3. FDA approves Roche's Zelboraf for rare blood cancer treatment
Recently, the US FDA expanded the indication of Roche drug Zelboraf (vemurafenib) for the treatment of adult patients with rare blood cancer Erdheim-Chester disease (ECD) and carrying BRAF V600 mutation, which is the first FDA approved ECD therapy.

ECD is a blood cancer that grows from the bone marrow and grows slowly. ECD promotes the proliferation of tissue cells (a type of white blood cells) that can cause tumors to invade multiple organs and tissues of the body, including the heart, lungs, and brain. It is estimated that ECD affects 600 to 700 patients worldwide, and their life expectancy is very limited, and new therapies are urgently needed to alleviate the disease.
Zelboraf is an oral small molecule kinase inhibitor that inhibits the growth of cancer cells by inhibiting the activity of B-raf proteins that promote cell growth. It was approved by the FDA in 2011 for the treatment of patients with unresectable or metastatic melanoma carrying the BRAF V600E mutation. About 54% of patients with ECD have BRAF V600 mutations, so researchers believe that Zelboraf is also expected to treat ECD. In response to this indication, the FDA has granted Zelboraf priority review qualifications, breakthrough therapy approvals, and orphan drug qualifications.
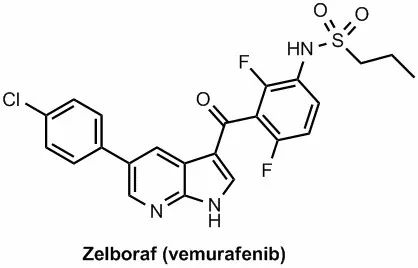
â–²The molecular structure of Vemurafenib (Source: C&EN)
Zelboraf's approval was based on its efficacy in a Phase 2 study of VE-BASKET. The trial included a total of 22 patients with ECF who were positive for BRAF-V600 mutations. The study measured the overall response rate (ORR) of the patients. The results showed that patients treated with Zelboraf had an ORR of 54.5%, of which 11 patients (50%) achieved partial remission and 1 patient (4.5%) achieved complete remission.
“Zelboraf is approved for ECD patients today, demonstrating how we apply knowledge of the underlying genetic traits of certain malignancies to other cancers,†said FDA Cancer Director, Office of Hematology and Oncology Products, FDA Center for Drug Evaluation and Research. Dr. Richard Pazdur, Director of the Centre of Excellence, said: "This product was first approved in 2011 for the treatment of certain melanoma patients with BRAF V600 mutations. We are now bringing this treatment to patients who have a rare cancer but have not been approved. â€
4. Genentech's new drug Alecensa is approved by the US FDA as first-line therapy for lung cancer
Genentech, a Roche company, today announced that the US FDA has approved Alecensa's (alectinib) new drug application (sNDA) for first-line treatment of metastatic lymphoma kinase (ALK)-positive metastatic non-small cells. Patients with lung cancer (NSCLC).

It is estimated that more than 220,000 people will be diagnosed with lung cancer in the United States in 2017, of which NSCLC accounts for 85% of all lung cancer. In the United States, approximately 5% of NSCLC patients are positive for ALK. This type of lung cancer is common in young people with a history of mild smoking or no smoking. These patients urgently need a targeted treatment to alleviate the disease.
Alecensa is one of the most promising lung cancer drugs that inhibits its signaling pathway by inhibiting the phosphorylation of ALK. Alecensa has been shown to inhibit a variety of mutant ALKs, some of which cause tumor cells to develop resistance to the existing standard therapy, crizotinib. In addition, Alecensa is able to cross the blood-brain barrier and enter the brain tissue and persist, which also has a significant killing effect on cancer cells that are transferred to brain tissue. Based on the excellent results of clinical trials, FDA has issued a breakthrough therapy certification and priority review for its applications for first-line treatment.
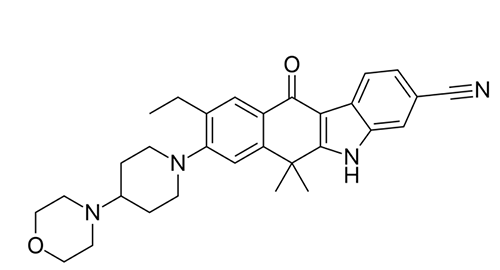
â–²Alectinib's molecular structure (Source: Wikipedia)
The approval is based on the results of a phase 3 clinical trial ALEX. This is an open-label, randomized, and actively controlled multicenter study evaluating the efficacy and safety of Alecensa in patients with ALK-positive NSCLC compared with crizotinib. They have not previously received systemic therapy for metastatic disease. . These patients were randomized to receive either Alecensa or crizotinib in a 1:1 ratio, with the primary efficacy endpoint being progression-free survival (PFS) as assessed by the investigator. Other efficacy endpoints included PFS, central nervous system (CNS) progression time, objective response rate (ORR), duration of remission (DOR), and overall survival (OS) as determined by an Independent Review Committee (IRC) assessment.
The results showed that Alecensa significantly reduced the risk of disease progression or death by 47% (HR=0.53, 95% CI: 0.38, 0.73, p<0.0001); the median PFS for patients treated with Alecensa was 25.7 months (95% CI: 19.9, not estimated), while the control group was 10.4 months (95% CI: 7.7, 14.6). The study also showed that Alecensa significantly reduced the risk of cancer spread in the brain or CNS by 84% (HR=0.16, 95% CI: 0.10, 0.28, p<0.0001). In addition, Alecensa's safety is consistent with previous research.
“Our goal is to develop drugs that significantly improve treatment standards,†said Dr. Sandra Horning, Chief Medical Officer and Head of Global Product Development at Genentech. “In our key study, Alecensa was significantly extended compared to crizotinib. People have no time for disease to worsen and show a significant reduction in the risk of cancer spreading to the brain."
5. Cellectis Universal CAR-T Therapy Receives FDA Release
Recently, Cellectis, which is committed to the development of allogeneic CAR-T therapy, announced that the US FDA has allowed its ongoing clinical trial of UCART123 to treat acute myeloid leukemia (AML) and blast cell-like dendritic cell tumors ( BPDCN).

UCART123 is a "universal" CAR-T therapy that differs from traditional CAR-T therapy in the source of cells: in traditional CAR-T therapy, researchers need to extract T cells from patients and insert chimeric antigens. The receptor (CAR) allows T cells to target specific antigens on cancer cells and then return these engineered T cells back to the patient for cancer treatment. Universal CAR-T therapy allows for the preparation of allogeneic T cells in advance and is readily available to patients. In addition, it is not affected by the quality of the patient's own T cells.
This therapy targets the CD123 antigen that is ubiquitously expressed on the surface of AML and BPDCN cancer cells. In February of this year, it became the first universal CAR-T therapy approved by the US FDA for clinical trials. Multiple patients were subsequently enrolled and treated. However, in this early clinical trial, one patient died and the US FDA decided to suspend the trial on September 4, so that it could be redesigned for safety. After discussions with the FDA, Cellectis decided to make the following changes to the current phase 1 clinical trial of UCART123:
Reduce the level of UCART123 cells to 62,500 per kilogram.
Reduce the amount of cyclophosphamide to 750 mg per square meter per day for 3 days. The maximum daily dose is no more than 1.33 grams.
Specific criteria were introduced on the day of UCART123 cell infusion, including the absence of new uncontrolled infections after lymphocyte clearance, no fever, only replacement doses of glucocorticoids, and no organ dysfunction.
Ensure that the next three patients in each treatment regimen are all under 65 years of age.
Ensuring that the next patient recruitment is a crossover between AML123 and ABC123, the patient recruitment interval for both studies should be no less than 28 days.
Currently, Cellectis is working with researchers and clinical trial centers to obtain approval from the Institutional Review Board (IRB) for revisions. If all goes well, Cellectis will resume the recruitment of patients and continue clinical trials.
6. ImmunoPulse IL-12 is expected to complement Cardutuda for the treatment of melanoma
OncoSec Medical recently announced positive results from a long-term follow-up of Phase 2 clinical trial OMS I-102. The trial evaluated the efficacy of ImmunoPulse IL-12 and Keytruda (pembrolizumab) in the treatment of melanoma patients who did not respond to anti-PD-1 therapy. This result will be announced at the annual meeting of the American Society for Cancer Immunotherapy (SITC).
Melanoma is a malignant tumor derived from melanocytes and is the most malignant cancer in skin cancer. Melanoma is easier to metastasize and spread. Once metastasis occurs, the patient's 5-year survival rate is only 4.6%. Although a number of immunotherapeutics have been approved for the treatment of advanced melanoma, only some patients will respond to these treatments and there are unmet medical needs.
OncoSec's immunotherapy platform, ImmunoPulse, specializes in providing IL-12, a natural protein with immunostimulatory properties. This therapy produces controlled localized expression of IL-12 in the tumor microenvironment, allowing the immune system to target and attack tumors in the body. This is expected to provide treatment options for those who do not respond to anti-PD-1 therapy. Currently, OncoSec is investigating ImmunoPulse IL-12 for the treatment of metastatic melanoma and triple-negative breast cancer.
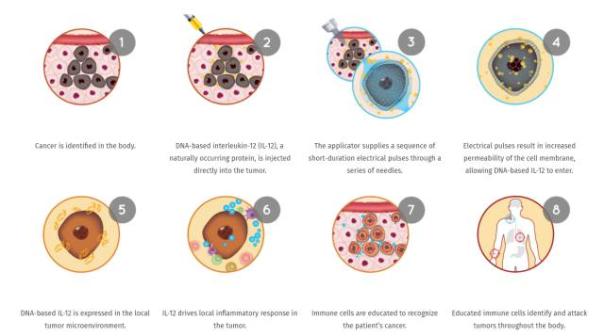
â–²ImmunoPulse IL-12 mechanism of action (Source: OncoSec Medical official website)
The updated data showed that progression-free survival (PFS) after combination of ImmunoPulse IL-12 plus pembrolizumab was 57% at 15 months, and the rate of remission was 100% (11/11). The median PFS has not yet been achieve. Based on a previously reported best overall response rate (BORR) of 50% and a complete response rate (CR) of 41%, the new data further demonstrate that this combination therapy can trigger a coordinated innate and adaptive immune response. And strongly suggests that it has a synergistic relationship with anti-PD-1 therapy. The latest findings further demonstrate that this combination therapy can reshape the tumor microenvironment, produce powerful intratumoral and systemic anti-tumor responses, convert "cold" tumors into "hot" tumors, and improve those that may not fight PD- 1 The clinical effect of a patient who responded to therapy.
"Stable PFS benefit and tolerability observed with ImmunoPulse IL-12 plus pembrolizumab is the first confirmed efficacy in a population of patients expected to be unresponsive to PD-1, and suggests that this combination therapy may represent the use of anti-PD-1 An important addition to the therapeutic field of metastatic melanoma patients after treatment," said Dr. Dan O'Connor, CEO of OncoSec.
Reference materials:
[1] FDA Sticks Breakthrough Tag on GlaxoSmithKline's Blood Cancer Drug
[2] Bristol-Myers Squibb Release: Opdivo (Nivolumab) Demonstrates Superior Three-Year Survival Benefit for Patients With Previously Treated Advanced Renal Cell Carcinoma (RCC)
[3] FDA approves first treatment for certain patients with Erdheim-Chester Disease, a rare blood cancer
[4] Genentech's Frontline Alecensa Approved for Non-Small Cell Lung Cancer
[5] FDA Lifts Clinical Hold on Cellectis Phase 1 Clinical Trials With UCART123 in AML and BPDCN
[6] OncoSec shares turn hot as microcap reports promising PhII data for an I/O one-two punch
Honey Sweet Baby Mandarin,Fresh Mandarin Orange,Baby Fresh Mandarin,Sweet Delicious Fresh Baby Mandarin
Laiwu Manhing Vegetables Fruits Corporation , https://www.manhingfood.com